
Hipercalcemia e linfadenopatia
Jovem com hipertensão e massa abdominal
Hipercalcemia e Febre em transplantado Renal
Hipotensão Durante Hemodiálise: uma etiologia rara que devemos lembrar!
IRA, fadiga e sedimento urinário ativo
Hipercalcemia por injeções cosméticas, como conduzir!
Estamos habituados a lidar com pacientes portadores de Doença Renal Crônica (DRC) que desenvolvem hiperfosfatemia, no entanto, como devemos abordar um quadro de hiperfosfatemia em pacientes com função renal não comprometida?
Neste artigo, publicado no Kidney International Reports ([link](https://www.kireports.org/action/showPdf?pii=S2468-0249%2823%2901525-5)) é relatado um caso clínico muito interessante.
O paciente em questão é um homem de 46 anos com histórico de osteomielite crônica desde os 20 anos. Já naquela época, ele apresentava um nível sérico de fósforo de 7,1 mg/dL, mantendo a função renal dentro da
normalidade.
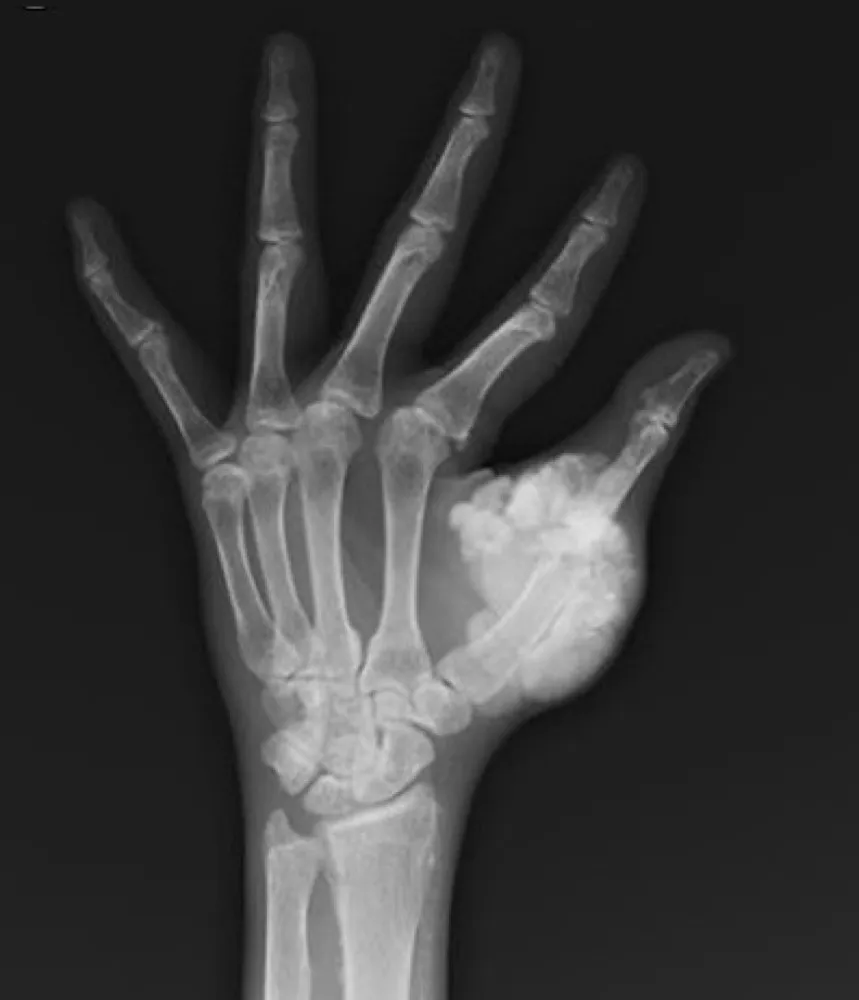
Na investigação de quadro de lesões de pele e calcificação extra-esquelética, observou-se um tumor calcificado no átrio esquerdo, bem como lesões ateroscleróticas calcificadas na aorta, artérias ilíacas e femorais, além de depósitos de cálcio no tecido subcutâneo e lesões na pele (figura abaixo).

Além disso, a investigação revelou uma baixa fração de excreção de fósforo, bem como níveis elevados de FGF23 (51 pg/mL, com o valor de referência <30 pg/mL) e C-terminal FGF23 (151 pg/mL, com o valor de referência <100 pg/mL).
Para investigar a fundo, realizou-se o sequenciamento de um painel com 358 genes associados a distúrbios esqueléticos, incluindo genes relacionados à homeostase do fósforo, tais como FGFR1, KLOTHO, FAM20, GALNT3 e
FGF23. Nesse sequenciamento, foram identificadas duas variantes missenses em heterozigose no gene GALNT3 (variantes: c.985G>A (p.G329R) e c.1312C>T (p.R438C)).
O paciente apresentava elevados níveis de fósforo, uma baixa fração de excreção de sódio e níveis elevados de FGF23. Do ponto de vista fisiológico, em resposta a uma sobrecarga de fósforo, ocorre a liberação de FGF23 e sua ligação ao receptor FGF-23/KLOTHO, resultando na redução da reabsorção de fósforo, promovendo assim a homeostase.
Mutação nos genes que interferem nessa homeostase do fósforo, como FGF23, GALNT3 e KLOTHO, podem criar o cenário clínico observado, ou seja, hiperfosfatemia com consequente deposição de cristais de fosfato de
cálcio e evolução para calcinose tumoral.
Geralmente, os pacientes afetados por essa condição mantêm níveis normais de cálcio, hormônio paratireoide (PTH) e vitamina D, no entanto, desenvolvem calcificações extensas fora do sistema esquelético, o que é uma
característica distintiva dessa condição.
Atualmente, não existe um tratamento específico para essa condição. O manejo atual envolve a implementação de uma dieta com baixo teor de fosfato (600–800 mg/d) e o uso de quelantes, como sevelamer ou hidróxido de
alumínio, sendo importante evitar o uso de quelantes de cálcio.
Pontos Importantes para o Aprendizado
* Desordens do metabolismo do fósforo são comuns em pacientes com DRC avançada.
* Hiperfosfatemia com função renal normal é uma condição extremamente rara e deve chamar a atenção para desordens genéticas subjacentes.
* Variantes genéticas no receptor FGF23 ou em seus reguladores podem levar à hiperfosfatemia e calcinose tumoral.
* O diagnóstico genético precoce e o manejo adequado podem ajudar a reduzir o risco de complicações vasculares nesses pacientes.